Phylometrics
 层序测量提供了一种新的数据挖掘方法来筛选提供的DNA序列
并使用强大的识别序列是重要的系统发育兴趣
分析工具. 被识别为与所提供的许多序列相似的序列
序列可以提供对它们的功能或进化关系的关键见解.
用户需要与浏览大多数互联网应用程序相同的基本计算机技能.
层序测量提供了一种新的数据挖掘方法来筛选提供的DNA序列
并使用强大的识别序列是重要的系统发育兴趣
分析工具. 被识别为与所提供的许多序列相似的序列
序列可以提供对它们的功能或进化关系的关键见解.
用户需要与浏览大多数互联网应用程序相同的基本计算机技能.
Summary
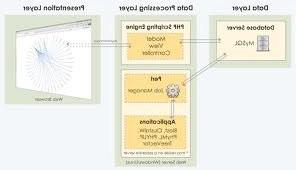
我们开发了一个网络应用,Phylometrics,它依赖于集体网络 BLASTN结果(而不是单个BLASTN命中)便于构建过程 系统发育树在自动化,高通量的方式,同时提供新颖的工具 寻找与最小人类有重要系统发育关系的序列 参与. 该应用程序可以在实验室本地安装或托管 远程使用简单的向导样式格式来引导用户通过管道 不需要编程背景. 此外,构造学实现了 一个独立的作业排队系统,使用户能够继续使用系统 而作业运行时性能几乎没有下降.
Background
比较序列分析16S rRNA基因是常用的特征 环境样品的微生物多样性. 然而,序列相似性确实如此 由于许多因素,并不总是意味着功能或进化上的相关性,包括 变化和收敛速度不等. 因此,依靠顶部BLASTN命中进行系统发育 研究可能会歪曲这些成分的多样性. 此外,尝试 通过在每个序列中包含大量BLASTN命中来规避这个问题 用一棵树来探索它们之间的关系会带来其他问题. 例如,倍数 如果不依赖于人工,序列对齐将会很差,并且计算成本很高 对齐,并且可能很难从结果中派生出有意义的关系 树. 然而,分析集体BLASTN结果中的序列关系网络, 揭示低等级但密切相关的序列.